Tamika Smith
Tamika Smith Tyrone Turner
Tyrone Turner
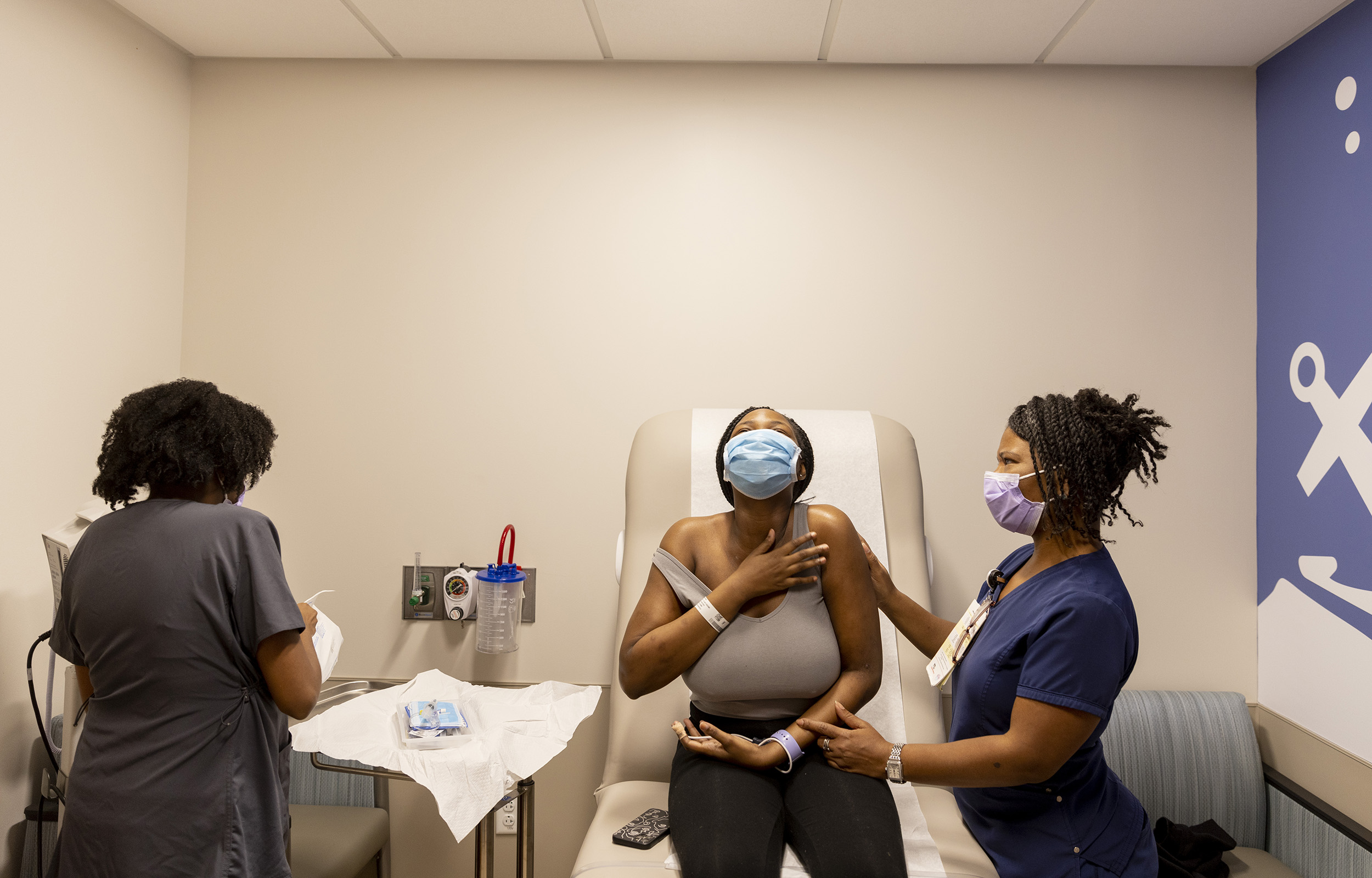
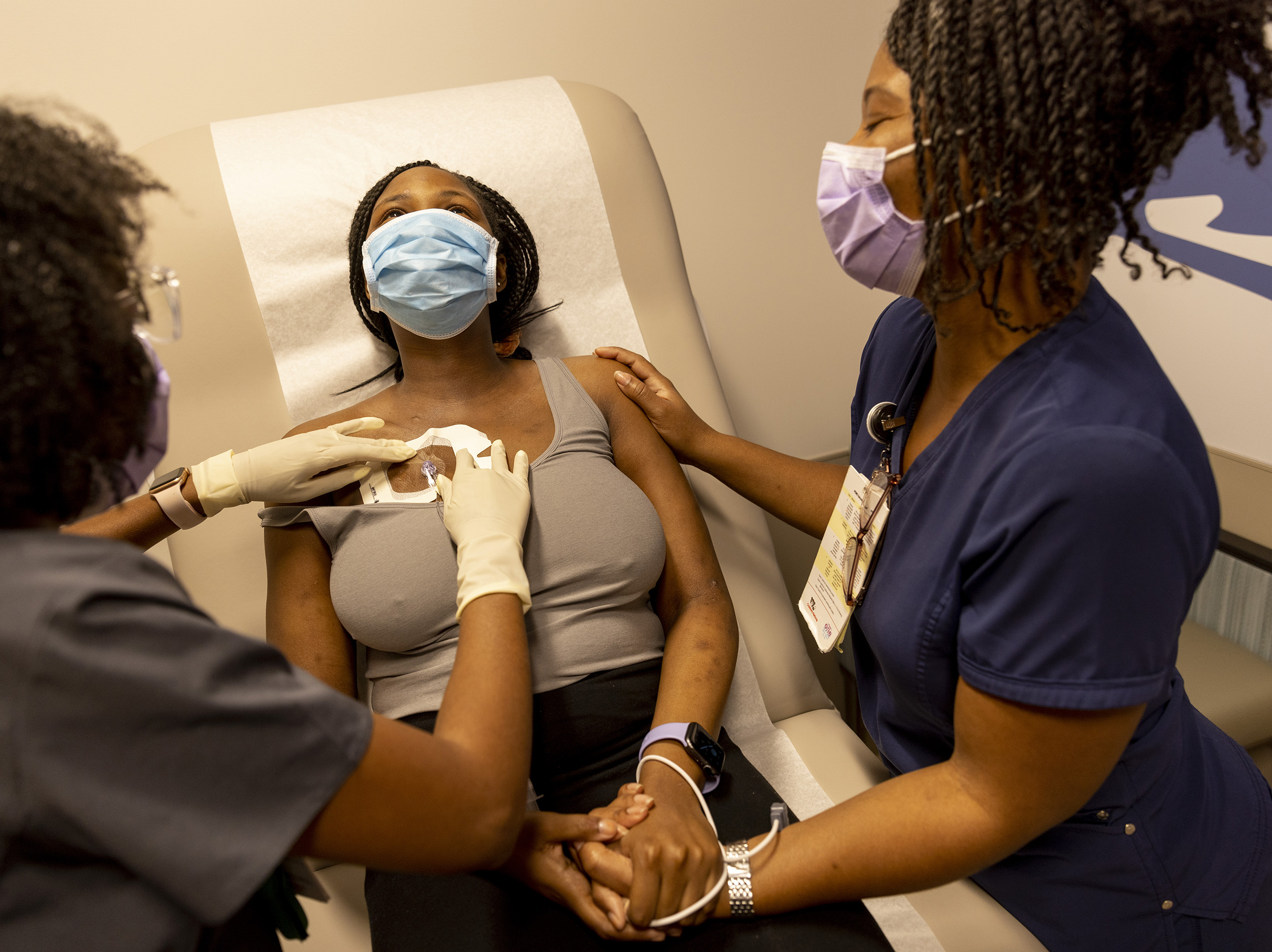
Kalani Sheffield is an energetic and ambitious 23-year-old who loves to bake, and aspires to be a nurse.
Standing in the middle of her kitchen in Prince George’s County, Maryland, she can’t help but smile when she talks about her potential career. But Sheffield’s joy quickly turns into a stream of tears when she speaks about a genetic blood disorder she’s living with – sickle cell disease – and how it puts her future in jeopardy.
Sheffield began her nursing studies at Trinity Washington University and had completed almost a year before periodic “crises”, or episodes of extreme pain due to her sickle cell, forced her to put her education on hold.
Sickle cell disease is a condition where red blood cells, which normally look like round discs, are shaped like sickles or crescent moons instead. They are also stiff, sticky, and get stuck together. This prevents blood from moving as it should, and can lead to pain and organ damage.



Dr. Stacey Fitzhugh, who studies sickle cell disease for the National Institutes of Health, says sickle cell disease can also lead to strokes and organ failure. The sickle cell crisis, or time of debilitating pain, is unpredictable and can sometimes last for weeks.
Sickle cell disease affects millions of people throughout the world and is most common among those whose ancestors came from sub-Saharan Africa or Spanish-speaking regions in the Western Hemisphere, according to research conducted by the CDC. In the United States, more than 100,000 people are affected with sickle cell, but it’s a disease that primarily affects the African American community, occurring in about 1 out of every 365 Black or African American births.

Kalani Sheffield’s brother Quadir, 24, has also been diagnosed with sickle cell disease. In fact, all four surviving Sheffield siblings are affected. Kalani Sheffield, her sister Zhari, 20, brothers Quadir and Nazi, 22, live with their mom and stepdad in their Prince George’s County home.

About 60 percent of the sickle cell population under the age of 21 followed at Children’s National Hospital in D.C. resides in Prince George’s County, says Dr. Andrew Campbell their lead hematologist, due to its proximity to their facility and their noteworthy expertise in treating the chronic ailment.
Samuel Price, 14, lives with his mother Annette Price in D.C. Price is much younger than the Sheffields and experiences sickle cell differently. “If you just imagine, like, somebody scratches you and they rip skin off, it’s similar to somebody scratching the skin off,” he says.

His crises aren’t as frequent, but can be very severe. Sometimes he develops acute chest syndrome, a condition in which the lungs can become filled with fluid, reducing oxygen levels, Dr. Campbell says.
When Samuel’s not being treated for sickle cell in the hospital, he says he’s focused on maintaining a healthy lifestyle. In the morning, he starts with his medications, “two hydroxyurea and one folic acid and sometimes I may have a flu pill in it,” he says.
Samuel knows that exercise is key to blood flow and circulation, so he goes on walks with his mom near Catholic University. Lately, he’s been excited about gaining endurance. “So, like, the first time we did it, I had to take three breaks, and the second time I did it, we only had to take one break.”


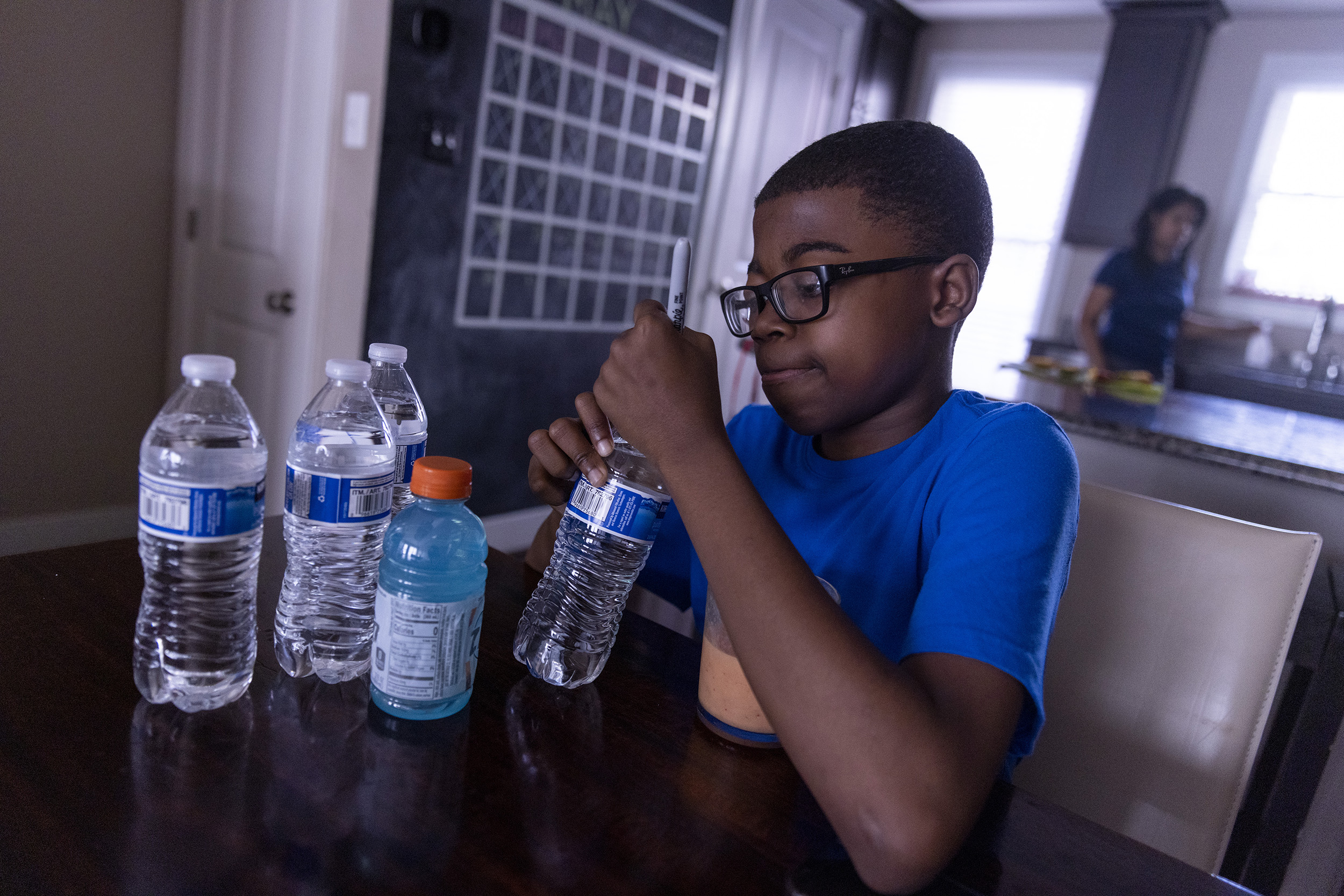
But all his precautions and preventive care only go so far – he can still go into a crisis at any time. Some specialists say age plays a contributing factor.
“I think the biology of the disease changes. There are changes that put people at more risk for developing chronic pain, which begins often in a patient in their twenties,” says Dr. Sophie Miriam Lanzkron, director for the Sickle Cell Center for Adults at Johns Hopkins Sickle Cell Center in Baltimore. “And, you know, there is an increased risk of mortality at this age.”
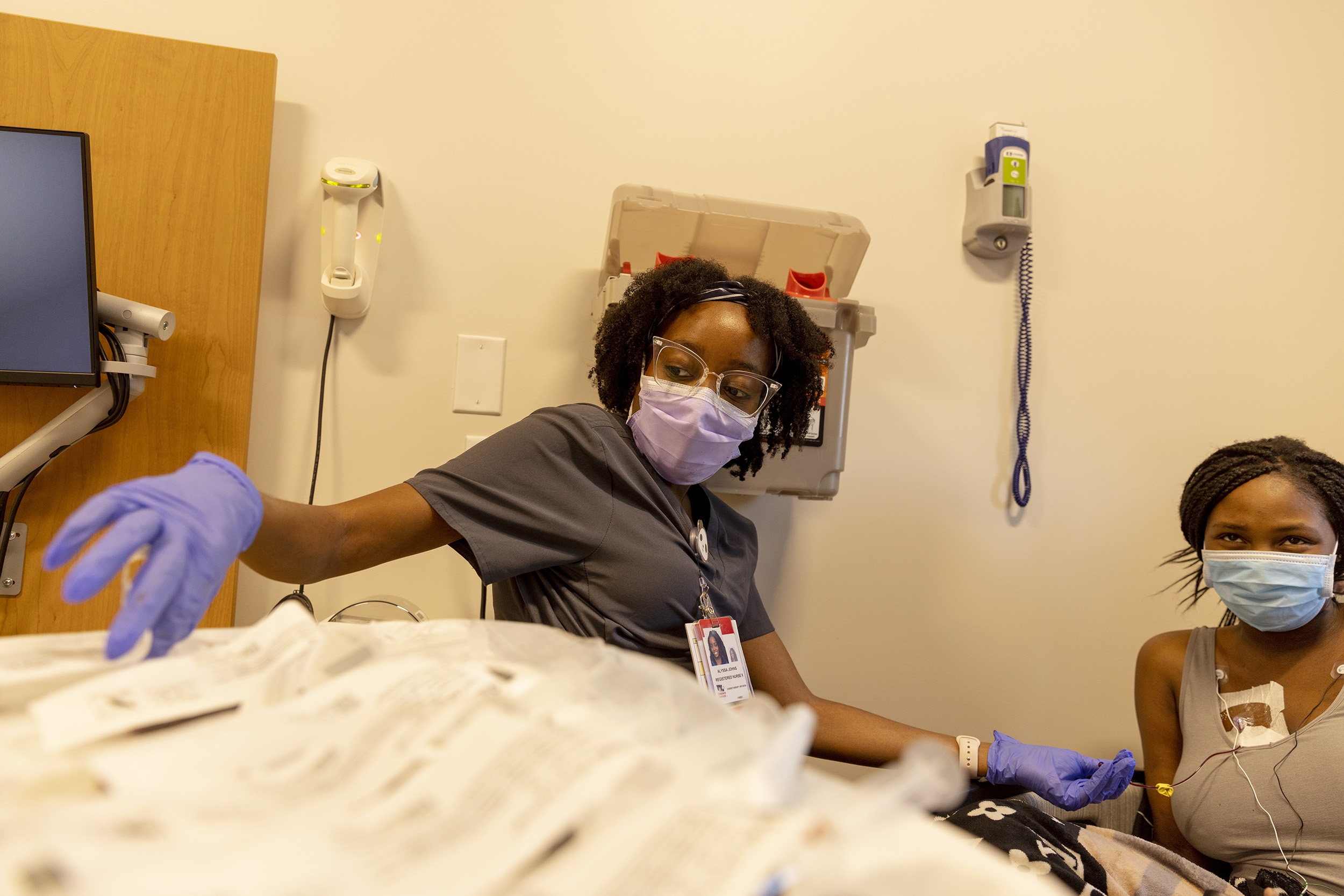
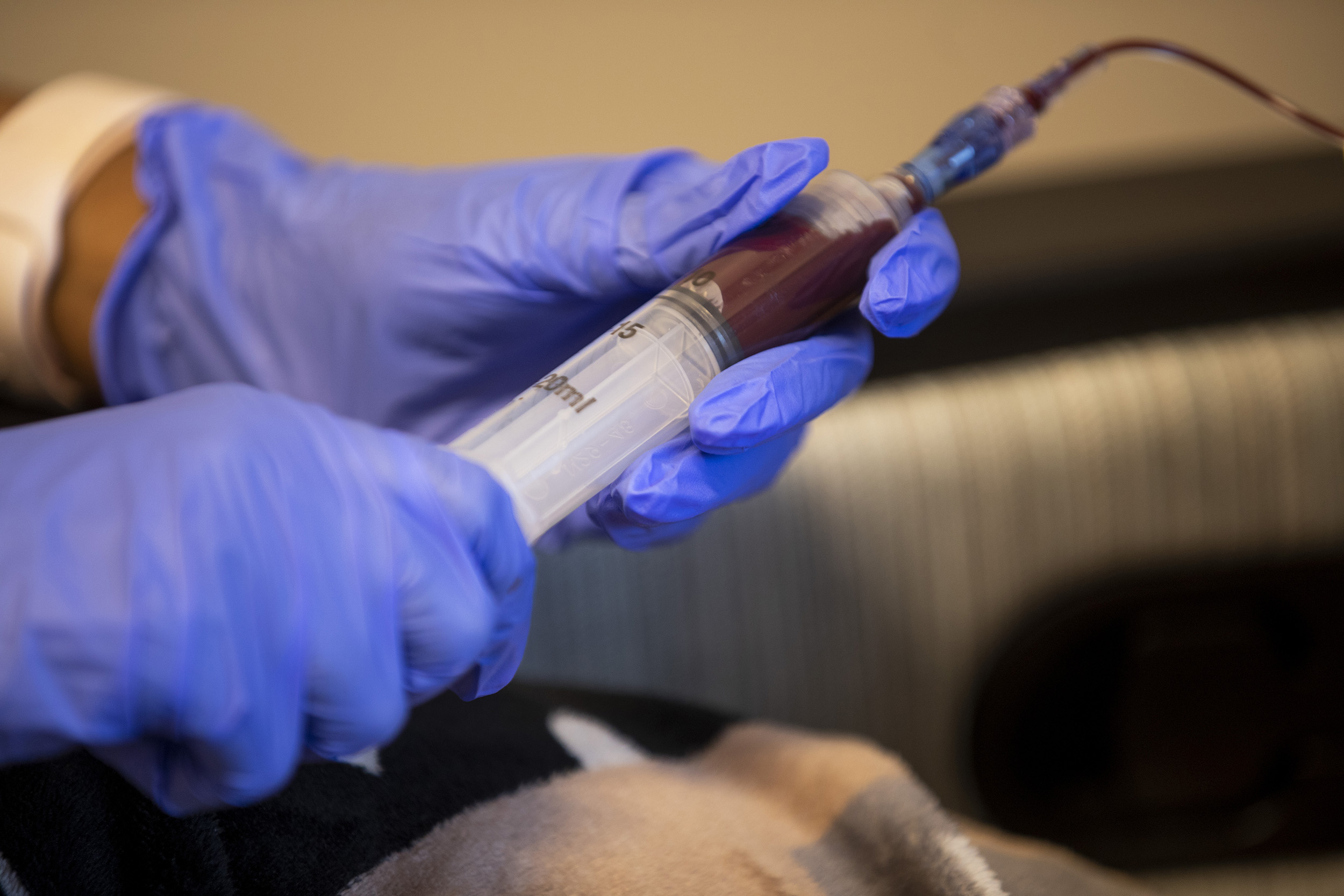
For many families managing sickle cell in the D.C. region, receiving treatment can be an insurmountable challenge to an already difficult situation, no matter what their age is.
There is the logistical and financial hurdle of traveling to appointments, some that can be as far as an hour away. “It’s a lot of money for these Ubers, these Lyfts, these rides,” Kalani Sheffield says. “You’re in pain. You’re not going to jump on a bus or train that’s affordable.”
Sheffield also says that, as she’s gotten older, her pain is treated with increasing callousness by doctors.

“At Children’s [Hospital] we go straight back [for treatment]. You’re a level one [patient],” she says. But once patients “age out”, treatment becomes different – and Sheffield says she’s often left waiting in the ER for extended periods of time, while in pain, waiting for treatment.
Riquel Richardson, 22, lives in Waldorf, Maryland and remembers when she started being treated differently in the ER.

“When I was younger, I felt like it was a lot easier. When I was younger, the staff was a lot nicer. They were a lot more caring,” she says.
Richardson commutes to Children’s National Hospital in D.C. from Waldorf, MD. It takes more than an hour to get there to receive the specialized care she needs. “And most of the hospitals I’ve been to [in Waldorf], doctors, they’ll ask me like, ‘Oh, what is sickle cell disease?’” she says.
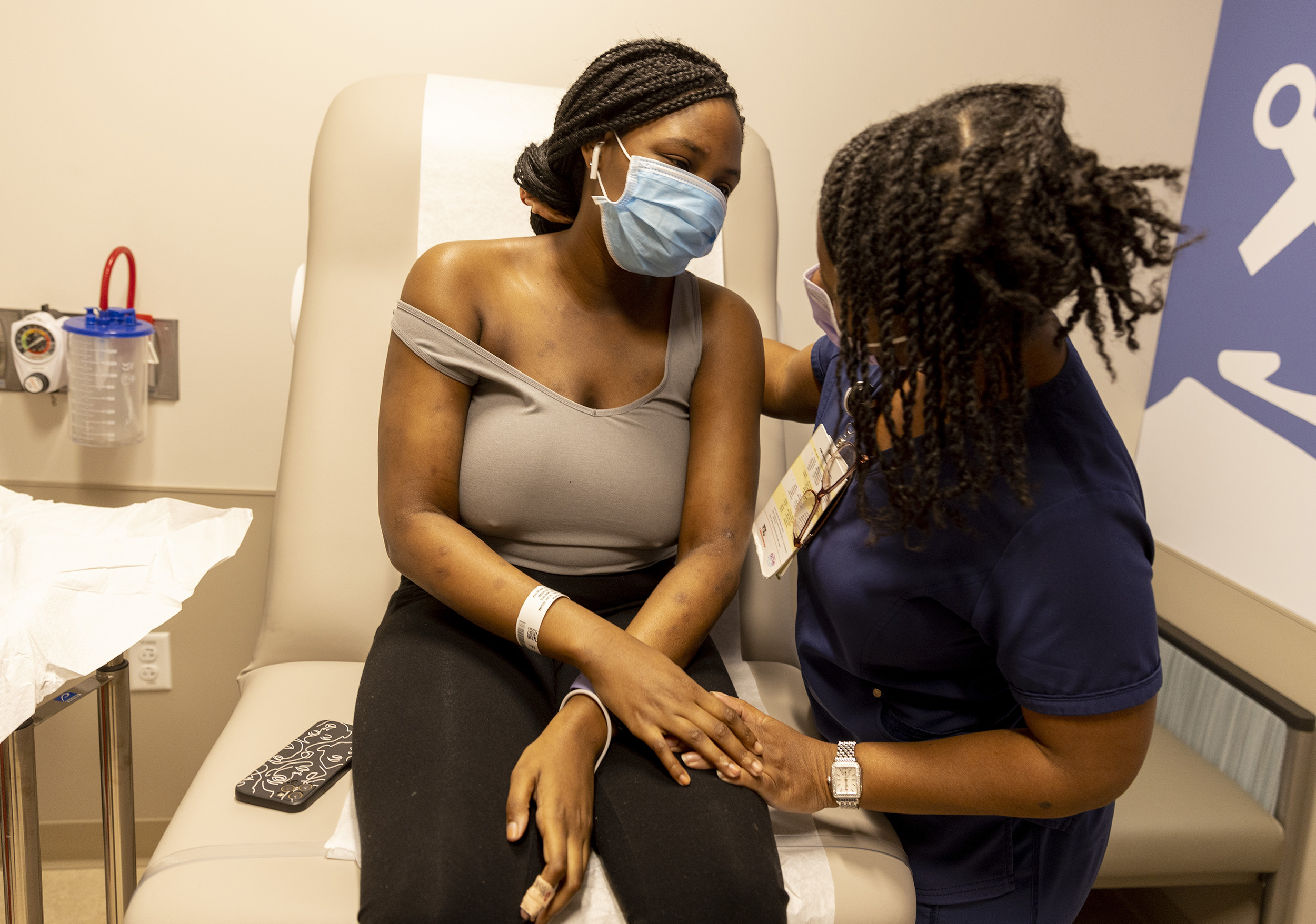
“And if I’m in a crisis, I don’t really have that much time to try and sit there and educate you and explain to you what it is that I’m going through,” she adds.
Quadir Sheffield, says he is often treated like a “drug seeker” when he goes to an ER for pain treatment during a sickle cell crisis.
Dr. Lanzkron says that opioids are most commonly diagnosed for the chronic pain that accompanies a sickle cell crisis. With multiple years of intake, drug tolerance increases and higher doses are needed to ease the pain. This can be off putting for medical professionals, she says, especially if they are unaware of the disorder.


One solution to this disconnect, says Dr. Lanzkron, could be patient specific treatment plans – which at this time isn’t the standard.
”There are lots of people who don’t have access to high quality sickle cell care, who do not have patient-specific treatment plans, who show up in an emergency department without any objective measure of crisis and are met with resistance,” Dr. Lanzkron says.
Since President Nixon signed the National Sickle Cell Anemia Control Act in 1972 providing funding for research, screening and counseling programs, and public awareness, the amount of people affected nationwide has more than doubled. However, resources for treating the disease, as well as finding a cure, have not multiplied, according to experts in the field, and there is a disparity between funding received for sickle cell and other comparable diseases.
For example, Dr. Lanzkron co-authored a study comparing funding for cystic fibrosis to that of sickle cell disease. They have lots in common: they are both genetic disorders that require lifelong comprehensive care and shorten life expectancy.
But between 2008 and 2018, sickle cell received about $800 of research funding per patient from the National Institutes of Health, while cystic fibrosis received $2,800 per patient.
What might explain that difference?
“The color of the skin of the people affected by the disease,” says Dr. Lanzkron. During the study period, about 90,000 mostly Black people were affected by sickle cell, while about 30,000 mostly white people were affected by cystic fibrosis.
“Now, can I tell you, that’s why the funding differences are so disparate? I can’t prove it, but it seems like it’s probably a big component of the problem,” Dr. Lanzkron says. “I know structural racism has had an enormous impact on the quality of care that people get. And so it would not be surprising that it also affects funding.”
Medical professionals and advocates in the sickle cell community recognize there is an issue with how people who are affected with the disease may be viewed within the healthcare system.
Derek and Shantá Robertson started the Maryland Sickle Cell Disease Association in 2006 because of their own experiences – two of their three sons have sickle cell disease. They were determined to create a comprehensive life plan for their sons, Khari Robertson, (now 24), and Mikaili Robertson, (now 22), and to educate the community around them.

The Robertsons developed a plan that included fostering a relationship with their local emergency room and their hematologists, and educating their son’s teachers and school nurses about their disease.
Derek Robertson is now working through his position with the Maryland Sickle Cell Disease Association to advocate for people who have sickle cell disease specifically in Prince George’s County.
Most recently, Derek Robertson, along with other community-based advocacy groups for sickle cell, collaborated with Dr. Lanzkron to apply for a $4 million grant from the Maryland Community Health Resources Commission, of which they were ultimately awarded about $2 million. These funds will be used to spearhead a comprehensive center for the treatment of sickle cell in Prince George’s County — a collaboration between Children’s National Hospital, Johns Hopkins, and University of Maryland Medical.
Dr. Stacey Little, vice president for Population Health and Community Health Lead at University of Maryland Medical Center, where the sickle cell center will be physically located, says the grant is “truly a blessing.”
”Right now, we know the average lifespan for a patient or individual with a sickle cell disease is about 40 years of age. And that is unconscionable in 2022,” says Dr. Little. She adds that the sickle cell center also plans to include a service site with a pain transfusion center, a medical provider for oversight, and a social worker. Dr. Little says her goal is to create, “a totally different pathway than what we have right now, a workflow” for sickle cell patients. The center is expected to open later this year, and offer additional services with time.
LaDawn Burnette, the mom of four kids with sickle cell disease, says she”appreciates the effort” but isn’t overly optimistic about the new comprehensive center.
Her thought is that it will leave her with a similar experience to when she visits Johns Hopkins Hospital in Baltimore. “The first thing they do is judge how much medication you’re on, the kids and I didn’t have a good experience at Johns Hopkins,” said Burnette.
Dr. Lanzkron says in her center prescribing medication for pain is a difficult balance to strike and sometimes it’s a difficult conversation to have with her patients.
“And the way we think about managing pain has really shifted for us in the adult sickle cell world. It used to be, if I saw a 22 year old who had chronic, everyday pain, I would write a script for a long acting opioid and say, ‘here you go.’”
She adds, “You’ve got a terrible disease that causes chronic pain. And we have to figure out how we’re going to manage that chronic pain without using chronic opioid therapy. Because I know if I put on chronic opioid therapy, I’m going to destroy your life.”
However it’s a decision Burnette says is not Dr. Lanzkron’s to make.
“Sickle cell is a chronic disease. At one time kids were not living past 16. My kids are in pain now and need relief now,” Burnette says.
Burnette’s daughter Kalani Sheffield, on the other hand, is eagerly awaiting the opening of the new sickle cell treatment center, saying that she’s “excited to see change within our medical facilities” and to see staff lead by ” science, facts and good healthcare and not by biases, stereotypes or discriminatory practices.”